Criterio. Revista Multidisciplinaria
Volumen 3 | No. 5 | Octubre 2023 - marzo 2024
https://revistacriterio.org
ISSN: 3006-2586 / ISSN-L: 3006-2586
http://doi.org/10.62319/criterio.v.2i5.20
Páginas 21 – 27

Técnicas para el análisis de poblaciones de la molécula de fibrinógeno
Techniques for the analysis of populations of the fibrinogen molecule
Marlin Angélica Guía Yanes
https://orcid.org/0000-0003-3924-4478
Universidad Central de Venezuela. Caracas, Venezuela
Artículo recibido 09 de marzo de 2023 / Arbitrado 25 de marzo de 2023 / Aceptado 18 mayo 2023 / Publicado 01 de octubre de 2023
RESUMEN
Este articulo tiene como propósito analizar la integridad del fibrinógeno purificado por electroforesis en un grupo de personas sanas para obtener un patrón de referencia considerado normal para hacer posible las comparaciones con los resultados provenientes de personas con enfermedad coronaria. Se realizó una investigación explicativa de identificación con un diseño experimental basado en la aplicación de técnicas de laboratorio para el análisis de varios tipos de poblaciones de la molécula de fibrinógeno en personas sanas. Se logró la purificación de la molécula por el método de Kazal (1963) con la posterior realización de electroforesis en gel de acrilamida SDS-PAGE (Sodium dodecyl sulfate polyacrylamide gel electrophoresis por la técnica de Laewmli (1970), obteniendo la visualización de las principales cadenas características de la molécula normal del fibrinógeno (α, β, y γ). Se concluye que la electroforesis en gel de acrilamida SDS-PAGE permite la visualización de las principales cadenas características de la molécula normal del fibrinógeno (α, β, y) para su posterior comparación con patrones patológicos.
Palabras clave: Fibrinógeno; molécula; proteína; análisis; electroforesis.
ABSTRACT
The purpose of this article is to analyze the integrity of fibrinogen purified by electrophoresis in a group of healthy people to obtain a reference standard considered normal to make comparisons possible with the results from people with coronary heart disease. An explanatory identification research was carried out with an experimental design based on the application of laboratory techniques for the analysis of various types of populations of the fibrinogen molecule in healthy people. The purification of the molecule was achieved by the method of Kazal (1963) with the subsequent performance of electrophoresis in acrylamide gel SDS-PAGE (Sodium dodecyl sulfate polyacrylamide gel electrophoresis by the technique of Laewmli (1970), obtaining the visualization of the main characteristic chains of the normal fibrinogen molecule (α, β, and γ). It is concluded that SDS-PAGE acrylamide gel electrophoresis allows the visualization of the main characteristic chains of the normal fibrinogen molecule (α, β, and) for later comparison with pathological patterns.
Keyword: Fibrinogen; molecule; protein; analysis; electrophoresis.
INTRODUCCIÓN
El fibrinógeno es una proteína plasmática que desempeña un papel fundamental en la hemostasia (factor I de la coagulación) y en otras actividades, como cicatrización de heridas y procesos inflamatorios, activándose como proteína de respuesta de fase aguda por lo que se incrementa en la inflamación. Es una glicoproteína que contiene aproximadamente 4% de carbohidratos (Lacoviello, et al., 2001; Producto Sigma-Aldrich, 2002), compuesta por 2964 aminoácidos, con un peso molecular de 340 kDa, se encuentra presente en el plasma en una concentración de 2-4 g/L, con un promedio de 2,8 g/L (Standeven et al., 2005; Tennet et al., 2007). La molécula del fibrinógeno está compuesta por 2 pares de cadenas polipeptídicas Aα, Bß y ɤ unidas entre sí por puentes disulfuro, con una estructura dimérica compuesta de dos subunidades idénticas. (Lowe et al., 2004).
La cadena Aα tiene 610 aminoácidos, peso molecular de 63,5 kDa, la cadena Bβ 461 aminoácidos con un peso molecular de 56 kDa y la cadena γ 411 aminoácidos y 47 kDa de peso molecular (Hirose et al., 1988; Gardlund et al., 1977). Los extremos amino terminales de las cadenas Aα y Bβ se les denomina fibrinopéptidos A y B, son las secuencias que corta la trombina durante la activación de la coagulación (Scheraga et al., 1957; Blombäck et al., 1978).
El fibrinógeno es una proteína plasmática sintetizada y ensamblada en el retículo endoplásmico de los hepatocitos o células parenquimatosas del hígado (Weisel, 2005). Su nivel puede aumentarse, de dos a diez veces, como parte de la respuesta de fase aguda con lesiones en los tejidos, infecciones e inflamación (Redman y Xia, 2001). Se ha encontrado relación entre el aumento de los niveles de fibrinógeno y el riesgo de enfermedad coronaria, un aumento en la porción de la cadena γ´ en pacientes con varias formas de enfermedad cardiovascular y en síndromes inflamatorios (Cooper et al., 2003; Siebenlist, et al., 2005; Wolfenstein-Todel y Mosesson, 1980).
Dada esta la relación existente entre el fibrinógeno y la enfermedad coronaria se plantea esta investigación con el propósito de aplicar técnicas para el análisis de la molécula de fibrinógeno en personas sanas, purificar la molécula de fibrinógeno por precipitación, analizar la integridad del fibrinógeno purificado por electroforesis en un grupo de personas sanas para obtener un patrón de referencia considerado normal y realizar comparaciones en estudios posteriores con los resultados derivados de los provenientes de personas con enfermedad coronaria.
MÉTODO
Se realizó una investigación explicativa de identificación con un diseño experimental basado en la aplicación de técnicas de laboratorio para el análisis de varios tipos de poblaciones de la molécula de fibrinógeno en personas sanas.
Se obtuvo muestras de 10 voluntarios evaluados medicamente como personas sanas que cumplen con los siguientes criterios de inclusión: femeninos o masculinos sin patologías heredadas/ adquiridas o historial de co-morbilidad asociada, cuyas edades oscilen entre 20-35 años con un índice de masa corporal 18,5-24,9 Kg/m2. Sin hábitos tabáquicos o alcohólicos y sin tratamiento farmacológico o ingesta de antiinflamatorios o AINES los últimos 7 días. Aceptaron participar en el estudio y firmaron el consentimiento informado.
Según Rice et al. (1999) se debe hacer de forma previa la asepsia y antisepsia de la zona de venopunción con alcohol 70%, se realiza la extracción de la sangre periférica de la vena cefálica o basílica ubicadas en la parte anterior del miembro superior según los criterios de recolección, transporte y almacenamiento de la muestra estandarizadas por la norma del National Comittee for Clinical Laboratory Standards.
Los primeros 3cc de sangre se descartaron para evitar la posible contaminación con factor tisular, luego se extrajeron 25 cc, se colocaron en tubos de centrifuga de 15 ml, que contienen citrato de sodio al 3,8% (129 mM) (Ritchie, 2016), en una proporción de 1:9 (1 volumen de anticoagulante para 9 volúmenes de sangre). Se realizó una primera centrifugación a 2000 g x 10min a 4°C, se extrajo el sobrenadante descartando el pellet, se realizó una segunda centrifugación a 2000 g x 10min a 4°C luego, el sobrenadante se separó en alícuotas y se conservó a -80°C.
RESULTADOS Y DISCUSIÓN
1. Purificación del fibrinógeno
Para la purificación de fibrinógeno se usó el método de Kazal que ha sido utilizado en numerosas investigaciones. La purificación se realizó en tres etapas: la adsorción, la precipitación y la eliminación del agente precipitante mediante la diálisis (Atlanta Biologicals, 2016).
Al plasma libre de plaquetas se añaden los inhibidores 1mM Pefabloc (4-(2-Aminoetil) benzenosulfonil fluor hidrochlorhidrico) y 5 mM EDTA Na2 5 mM (ácido etilendiaminotetraacético).
Si la muestra se mantuvo a -80 °C, los inhibidores se añaden antes de descongelar, si está a 37°C se añaden y se procede a eliminar los factores vitamina K dependientes por adsorción añadiendo 20 mM de sulfato de magnesio (MgSO4) y 90 gr/L de sulfato de bario (BaSO4) dejando la muestra en rotación de 360º durante 30 min. Luego se centrifugó a 2000 g x 15 min a 22°C. Se tomó el plasma sobrenadante y se realizó una segunda adsorción.
Al plasma adsorbido y en agitación continúa evitando la formación de burbujas, se le añadió lentamente la glicina a una concentración final de 2,2 M. El fibrinógeno precipitado por glicina se separó centrifugando a 2000 g x 30 min a 22 °C. Se descartó el sobrenadante y el precipitado se resuspendió mediante agitación por 30 min en tampón Tris en un volumen igual al plasma inicial.
La precipitación se realizó tres veces de la misma manera para la obtención de un fibrinógeno parcialmente purificado. La tercera precipitación se resuspendió en un ¼ del volumen del plasma inicial y se procedió a dializar la muestra durante toda la noche en tampón Tris 50 mM, NaCl 0,15 M a pH 7,4.
Luego de la obtención del fibrinógeno purificado se midió la concentración de este empleando el coeficiente de extinción molar del fibrinógeno a 280 nm (1,51ml/mg x cm) utilizando de blanco Tris – NaCl salino.
2. Electroforesis SDS-PAGE
Es un método de cuantificación, composición y caracterización de las proteínas. Previo a la electroforesis es necesario determinar el porcentaje de proteína contenido en la muestra, usualmente se carga de 10 a 40 µg de proteína total por carril, sometiendo al gel contenido con las muestras a un campo eléctrico con el denominado tampón de electroforesis. Se usaron 10 µl de plasma de los cuales se tomó la cantidad de µl requerida en base a los mg/ µl de Fg de la muestra por 5 mg, con Tris 6,8% y Buffer urea SDS. Se trató de la técnica discontinua de Laewmli descrita por Garfin (1990) en su investigación.
Se prepararon las soluciones según los gramos indicados para el porcentaje del gel, se preparan las campanas con las láminas y los separadores, se coloca entre las dos laminas la preparación del gel y se espera que ocurra la polimerización del gel de resolución, luego se preparó el stacking gel y se esperó su polimerización para realizar la carga de los pocillos con las muestras y comenzar la corrida a 80 v por una hora y posterior 120 por media hora.
Gráfico 1. Resultados obtenidos de la molécula de fibrinógeno en Electroforesis SDS-PAGE en gel al 8%
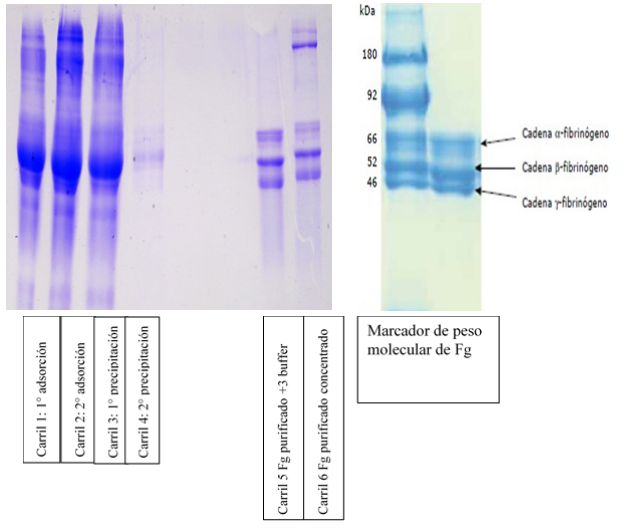
Una vez conseguido el gel se compara con un marcador de peso molecular para fibrinógeno humano obteniendo el patrón normal con el cual se compararán las muestras provenientes de personas con enfermedad coronaria.
En los carriles 5 y 6 correspondientes a la muestra final de fibrinógeno purificado se pueden apreciar la presencia de las tres cadenas (alfa, beta y gamma) que componen la molécula, comparadas con un patrón comercial de peso molecular normal para el fibrinógeno.
Método de purificación de Kazal
El método de purificación de fibrinógeno de Kazal se puede resumir en tres etapas esenciales: la adsorción, la precipitación y la diálisis. Al plasma libre de plaquetas (10 ml) se añaden inhibidores de proteasas (1mM Pefabloc (4-(2-Aminoetil) benzenosulfonil fluor hidroclorhidico) y 5 mM EDTA Na2 (ácido etilendiaminotetraacétato disódico) (Atlanta Biologicals, 2016)
El Pefabloc es usado para inhibir las proteasas las cuales pudieran degradar a la proteína a purificar. Las proteasas son omnipresentes en todas las células vivas, tan pronto como las células se lisan, las proteasas son liberadas y puede degradar rápidamente cualquier proteína. Esto puede reducir drásticamente el rendimiento de la proteína y la purificación (Johnson, 2016).
El EDTA Na2 (ácido etilendiaminotetraacético) actúa como agente quelante del calcio, lo que impide la activación de la coagulación. Es un ácido que contiene cuatro grupos ácido carboxílico y dos grupos amino con electrones de pares solitarios que quelan el calcio y otros iones metálicos (Kazal et al., 1963).
Se procede a adsorber los factores vitamina K dependientes según la técnica descrita por Banfi et al. (2007).
Se agrega al plasma 20 mM de sulfato de magnesio (MgSO4) y 90 gr/L de sulfato de bario (BaSO4) dejando los tubos rotando durante 30 min. Se centrifuga a 2000 g x 15 min a 22°C, se toma el sobrenadante y se realiza una segunda absorción.
Al plasma adsorbido y en agitación continúa evitando la formación de burbujas, se le añade lentamente la glicina a una concentración final de 2,2 M. El fibrinógeno precipitado por glicina se separa centrifugando a 2000 g x 30 min a 22 °C. Se descarta el sobrenadante y el precipitado se resuspende mediante agitación por 30 min en tampón Tris en un volumen igual al plasma inicial.
La precipitación se realiza tres veces de la misma manera para la obtención de un fibrinógeno parcialmente purificado. A través de este método se consigue purificar la proteína de fibrinógeno humano para aislar y caracterizar la molécula en estudio.
Electroforesis en gel de acrilamida SDS-PAGE (Sodium dodecyl sulfate polyacrylamide gel electrophoresis:
La electroforesis se ha desarrollado extensamente para las separaciones moleculares, el principio se basa en mover moléculas cargadas por solución aplicando un campo eléctrico a través de la cual se pasa la mezcla con las moléculas en movimiento en un campo eléctrico con una velocidad dependiente de su carga, forma y tamaño (Bradley y Markwell, 2007). El análisis SDS-PAGE es la herramienta de análisis de purificación de proteínas dominante, debido a su simplicidad y capacidad genérica para separar y detectar proteínas. Se utiliza para supervisar y verificar el progreso de una purificación.
Por medio de la electroforesis se separan proteínas con una masa molecular relativa no inferior a 10 kD. Las proteínas muy pequeñas (<10 kD) son difíciles de resolver debido a su baja capacidad de unión al SDS, que puede resolverse mediante geles de gradiente o utilizando diferentes condiciones de electroforesis (Garfin, 1990). El principio de la separación es que las proteínas migran a través de poros progresivamente más pequeños que oponen resistencia al paso de las moléculas, este permite a las proteínas más pequeñas viajar más rápidamente que las proteínas más grandes y finalmente tienden a detenerse y concentrarse donde el poro tamaño es demasiado pequeño para permitir una mayor migración (Casas-Terradellas, 2010). Los tamaños de los poros están regulados por la concentración de gel el cual disminuye con el incremento del porcentaje en la concentración de acrilamida (% T) con lo que la separación se produce a velocidades diferentes en función del tiempo. La bys-acrilamida puede actuar como terminador de la cadena en el proceso de polimerización y las concentraciones altas pueden disminuir el tamaño de poro máximo de gel (Gill & Von Hippel, 1989). Para mejorar la nitidez de la banda y separar las proteínas a analizar por electroforesis el gel resolutivo se corona con una acrilamida de menor porcentaje y diferente pH llamado Stacking gel o gel de apilamiento (Garfin, 1990). Una vez obtenido el gel que caracteriza la molécula de fibrinógeno se compara con el patrón pre establecido de peso molecular dado para el fibrinógeno en personas sanas, obteniendo las tres cadenas (α, β, y γ), en pacientes con enfermedad coronaria se ha documentado la existencia de variantes con respecto a este patrón considerado como normal, en el que se ha observado la presencia de variaciones ><10 kD) son difíciles de resolver debido a su baja capacidad de unión al SDS, que puede resolverse mediante geles de gradiente o utilizando diferentes condiciones de electroforesis (Garfin, 1990).
El principio de la separación es que las proteínas migran a través de poros progresivamente más pequeños que oponen resistencia al paso de las moléculas, este permite a las proteínas más pequeñas viajar más rápidamente que las proteínas más grandes y finalmente tienden a detenerse y concentrarse donde el poro tamaño es demasiado pequeño para permitir una mayor migración (Casas-Terradellas, 2010).
Los tamaños de los poros están regulados por la concentración de gel el cual disminuye con el incremento del porcentaje en la concentración de acrilamida (% T) con lo que la separación se produce a velocidades diferentes en función del tiempo. La bys-acrilamida puede actuar como terminador de la cadena en el proceso de polimerización y las concentraciones altas pueden disminuir el tamaño de poro máximo de gel (Gill & Von Hippel, 1989).
Para mejorar la nitidez de la banda y separar las proteínas a analizar por electroforesis el gel resolutivo se corona con una acrilamida de menor porcentaje y diferente pH llamado Stacking gel o gel de apilamiento (Garfin, 1990).
Una vez obtenido el gel que caracteriza la molécula de fibrinógeno se compara con el patrón preestablecido de peso molecular dado para el fibrinógeno en personas sanas, obteniendo las tres cadenas (α, β, y γ), en pacientes con enfermedad coronaria se ha documentado la existencia de variantes con respecto a este patrón considerado como normal, en el que se ha observado la presencia de variaciones que muestran la heterogeneidad de las cadenas, en un mayor porcentaje con la aparición de la variante γ´ en este tipo de pacientes.
CONCLUSIONES
La aplicación de las distintas técnicas para el análisis de las poblaciones de las moléculas de fibrinógeno permite obtener patrones de referencias de individuos sanos realizados en el laboratorio, por el investigador basado en técnicas ya conocidas con los cuales se contrastarán los resultados de muestras provenientes de personas con enfermedad coronaria.
En los resultados comparados y coincidentes con el marcador de peso molecular comercial, se obtuvo un patrón normal de la molécula de fibrinógeno a través de métodos que permiten su repetibilidad y reproducibilidad con los mismos resultados en sujetos sanos.
Esto va a permitir establecer comparaciones con muestras de fibrinógenos de pacientes con problemas coronarios para determinar la modificación molecular producida en esta molécula a partir de eventos patológicos en el organismo.
REFERENCIAS
Atlanta Biologicals. (2016). HEPES Buffer Solution (1M). https://www.atlanta-biologicals.com/shop/reagents-and-supplements/hepes-buffer-solution-1-m/
Banfi, G., Salvagno G.L., Lippi, G. (2007). The role of ethylenediamine tetraacetic acid (EDTA) as in vitro anticoagulant for diagnostic purposes. Clin Chem Lab Med; 45(5): 565 -76.
Blombäck B. Hessel B. Hogg D, A (1978). two-step fibrinogen-fibrin transition in blood coagulation. Nature; 275: 501–505.
Bradley, J.S., Markwell, J. (2007). Assay for determination of protein concentration. Current protocols in protein science. DOI 10.1002/0471141755.pha03as38
Casas-Terradellas E. Garcia-Gonzalo F. Hadjebi Q. Bartrons R. Ventura F. Rosa J. (2010). Simultaneous electrophoretic analysis of proteins of very high and low molecular weights using low-percentage acrylamide gel and a gradient SDS-PAGE gel. Electrophoresis; 31(8): 1318 - 1321.
Cooper A, Standeven K y Ariëns. (2003). Fibrinogen gamma-chain splice variant ɤ´alters fibrin formation and structure. Blood; 102(2): 535 – 540.
Gardlund B, Hessel B, Marguerie G, Murano G, Blomback B. (1977). Primary Structure of Human Fibrinogen Characterization of Disulfide-Containing Cyanogen-Bromide Fragments. Eur. J. Biochem. (77): 595 – 610.
Garfin, D. E. (1990). One dimensional gel electrophoresis Chapter Twenty-nine. https://www.researchgate.net/.../Chapter+29.+One-Dimensional+Gel+electrophoresis
Gill, S. & Von Hippel, P. (1989). Calculation of Protein Extinction Coefficients from Amino Acid Sequence Data. Analytical Biochemistry; 182: 319 – 326.
Hirose, S. Oda K. Ikehara Y. (1988). Biosíntesis, ensamblaje y secreción de fibrinógeno en hepatocitos de rata cultivados. Biochem. J.251: 373 – 377.
Johnson, M. (2016). Detergents: Triton X-100, Tween-20. https://www.labome.com/method/DetergentsTriton-X-100-Tween-20-and-More.html
Kazal, LA, Amsel, S., Miller, OP y Tocantins, LM (1963). La preparación y algunas propiedades del fibrinógeno precipitado del plasma humano mediante glicina. Actas de la Sociedad de Biología y Medicina Experimentales, 113 (4), 989-994.
Lacoviello, L. Vischetti, M. Zito, F. Benedetta Donati M. (2001). Los genes que codifican el fibrinógeno y el riesgo cardiovascular. Hipertensión. 38: 1199–1203. Laemmli-SDS-PAGE. Bio-protocol 1970. DOI: bio-protocol.org/bio101/e80
Lowe G, Rumley A y Mackie I. (2004). Plasma fibrinogen. Ann Clin Biochem; (41): 430–440.
Producto Sigma-Aldrich. (2002). Fibrinógeno de plasma humano. Número F4883. https://www.sigmaaldrich.com/catalog/product/sigma/f4883?lang=en®ion=VE
Redman, C., Xia, H. (2001). Ensamblaje de Biosíntesis de Fibrinógeno, Degradación Intracelular y Asociación con Síntesis y Secreción de Lípidos Ann N Y Acad Sci. 936:480 - 95.
Rice S. Browning C. Burke J. Nguten L. (1999). Membrane loader for gel electrophoresis. Genetic Biosystems. http://www.freepatentsonline.com/5972188.pdf
Ritchie, C. (2016). Purificatión protein. Labome Laboratories. http://www.labome.fr/method/ProteinPurification.html
Robert, L. Brumley, Jr.; Lloyd, M. (1993). Horizontal gel electrophoresis United States Patent
Scheraga H. Laskowski M Jr. (1957). The fibrinogen-fibrin conversion. Adv Protein Chem (12): 1–19.
Siebenlist, K. R., Mosesson, M. W., Hernandez, I., Bush, L. A., Di Cera, E., Shainoff, J. R., ... & Stojanovic, L. (2005). Studies on the basis for the properties of fibrin produced from fibrinogen-containing γ′ chains. Blood, 106(8), 2730-2736.
Standeven K. Ariens, R. Grant, P. (2005). La fisiología molecular y la patología de la fibrina/ función de sangre. Blood Rev. 19 (5): 275-288. Tennet G. Brennan S. Stangou A. Grady J., Hawkins P y Pepys M. (2007). El fibrinógeno plasmático humano se sintetiza en el hígado. Blood. 109(5): 1971-1974.
Weisel J. W. (2005). Fibrinogen and fibrin. Adv Protein Chem; (70): 247–299.
Wolfenstein-Todel, C. y Mosesson, M.W. (1980). Heterogeneidad del fibrinógeno plasmático humano: evidencia de una secuencia carboxilo terminal extendida en una variante normal de la cadena gamma (gamma’). Actas de la Academia Nacional de Ciencias, 77 (9), 5069-5073.